Introduction
For decades, sickle cell disease and β-thalassemia have been the inherited shadows of hemoglobin — molecular errors in the script of life that condemned millions to chronic pain, transfusions, and early mortality. Their management was palliative, not curative. Then, in 2023, the story changed. The United Kingdom’s Medicines and Healthcare products Regulatory Agency (MHRA) approved Casgevy (exagamglogene autotemcel) — the world’s first CRISPR-Cas9–based gene-editing therapy. Within months, the U.S. Food and Drug Administration and the European Medicines Agency followed. For the first time, the idea of rewriting—not just reading—the human genome reached the clinic.1–3
How Casgevy Works
At the molecular level, Casgevy uses the precision of CRISPR-Cas9 to target a genetic switch that controls fetal hemoglobin (HbF). Scientists harvest a patient’s own hematopoietic stem and progenitor cells (HSPCs) then outside the body, use CRISPR to disrupt the erythroid-specific enhancer of the BCL11A gene — a master repressor of HbF. This disruption reawakens a silenced genetic program from infancy: the fetal hemoglobin pathway.4,5
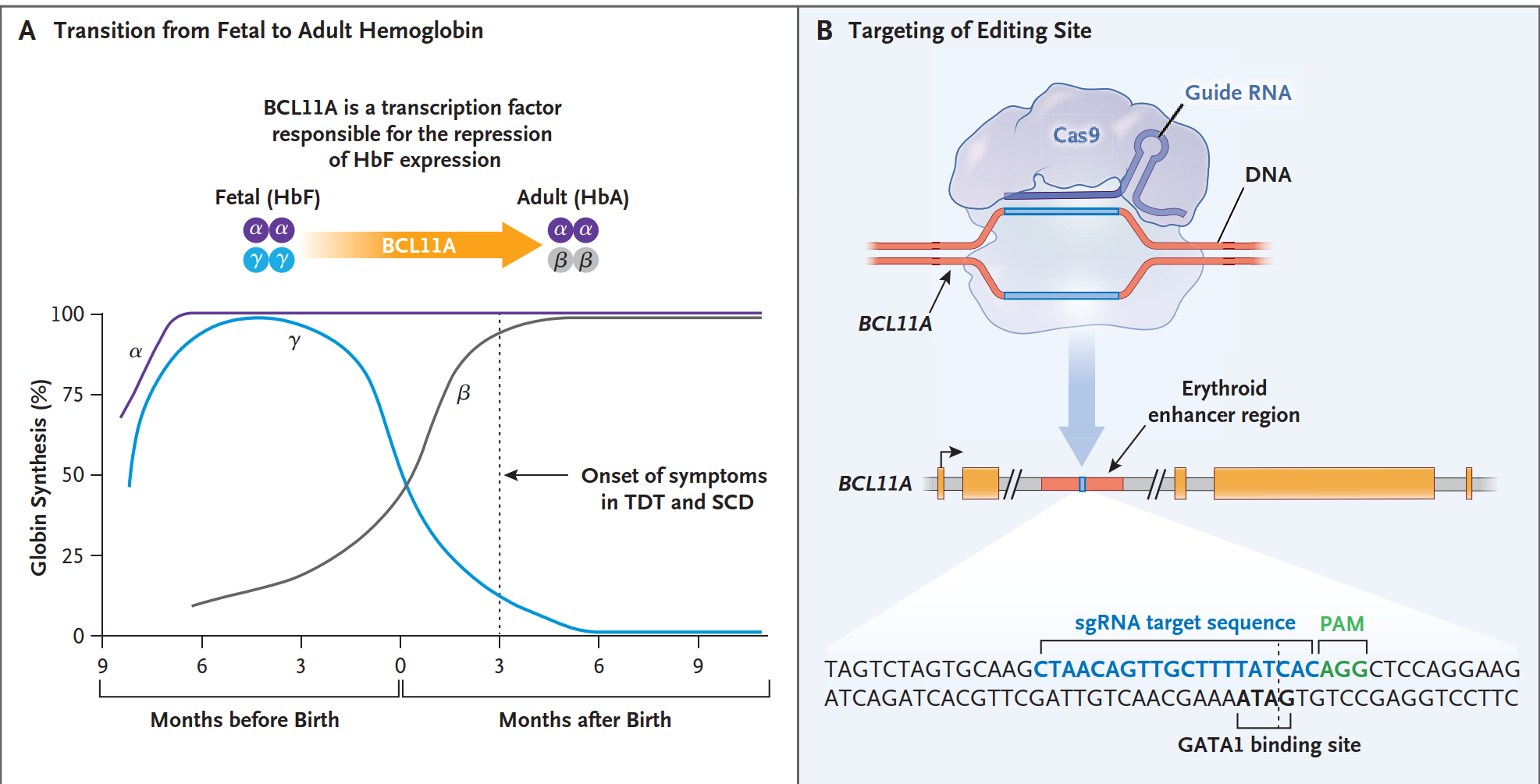
In sickle cell disease (SCD), the reactivated HbF prevents sickle hemoglobin (HbS) from polymerizing, halting the cascade of red-cell deformation and vaso-occlusion.6
In transfusion-dependent β-thalassemia (TDT), increased HbF compensates for the missing or defective adult hemoglobin (HbA), restoring effective oxygen transport.6
It is a quiet revolution at the chromosomal level — a single edit transforming lifelong disease biology.
Manufacturing and Infusion Process
The treatment protocol unfold as follow:
- Stem Cell Mobilization and Collection:
Patients receive agents (plerixafor and filgrastim) that draw stem cells from bone marrow into the bloodstream, followed by apheresis to collect CD34⁺ cells.
Plerixafor (AMD3100)
Plerixafor (AMD3100) is a selective inhibitor of CXCR4 , a chemokine receptor essential for retaining stem cells inside the bone-marrow niche.
Mechanism of Action
Plerixafor blocks the interaction between CXCR4 on hematopoietic stem cells SDF-1 (CXCL12) produced by marrow stromal cells
This CXCR4–SDF-1 axis acts as a “biological anchor” holding stem cells in the marrow.
When plerixafor inhibits this pathway:
HSPCs rapidly detach from their stromal microenvironment
They migrate into the bloodstream within hours
Mobilization is synergistically enhanced when combined with filgrastim
Plerixafor is typically given after several days of filgrastim, just before apheresis.
Filgrastim (G-CSF)
Filgrastim is a recombinant form of granulocyte colony-stimulating factor (G-CSF), a cytokine that plays a key role in neutrophil production and stem-cell kinetics.
Mechanism of Action
Filgrastim binds to the G-CSF receptor on myeloid progenitor cells, triggering:
Proliferation and differentiation of neutrophil precursors
Enhanced release of hematopoietic stem and progenitor cells (HSPCs) into the bloodstream
Improved survival and function of mature neutrophils
Why Filgrastim Mobilizes Stem Cells
Under high-dose stimulation, G-CSF causes:
Disruption of marrow adhesion signals (CXCR4–SDF-1 axis)
Increased protease activity (MMP-9, neutrophil elastase)
Decreased stromal retention of stem cells
This shifts HSPCs from the marrow niche into peripheral blood, where they can be collected via apheresis.
Filgrastim is commonly given daily for several days before stem-cell harvest
- Ex Vivo Gene Editing
In a controlled lab environment, CRISPR-Cas9 precisely edits the BCL11A enhancer within these cells.
- Conditioning
Before reinfusion, patients undergo myeloablative therapy with busulfan — a necessary reset that clears marrow space for the edited cells to engraft.
- Reinfusion
The modified stem cells are returned intravenously, homing back to the marrow where they begin producing HbF-rich red cells.
- Recovery and Monitoring
Intensive inpatient monitoring follows, often within specialized transplant centers capable of managing infection, cytopenia, and engraftment risks.
The therapy is delivered once — but its effects may last a lifetime.
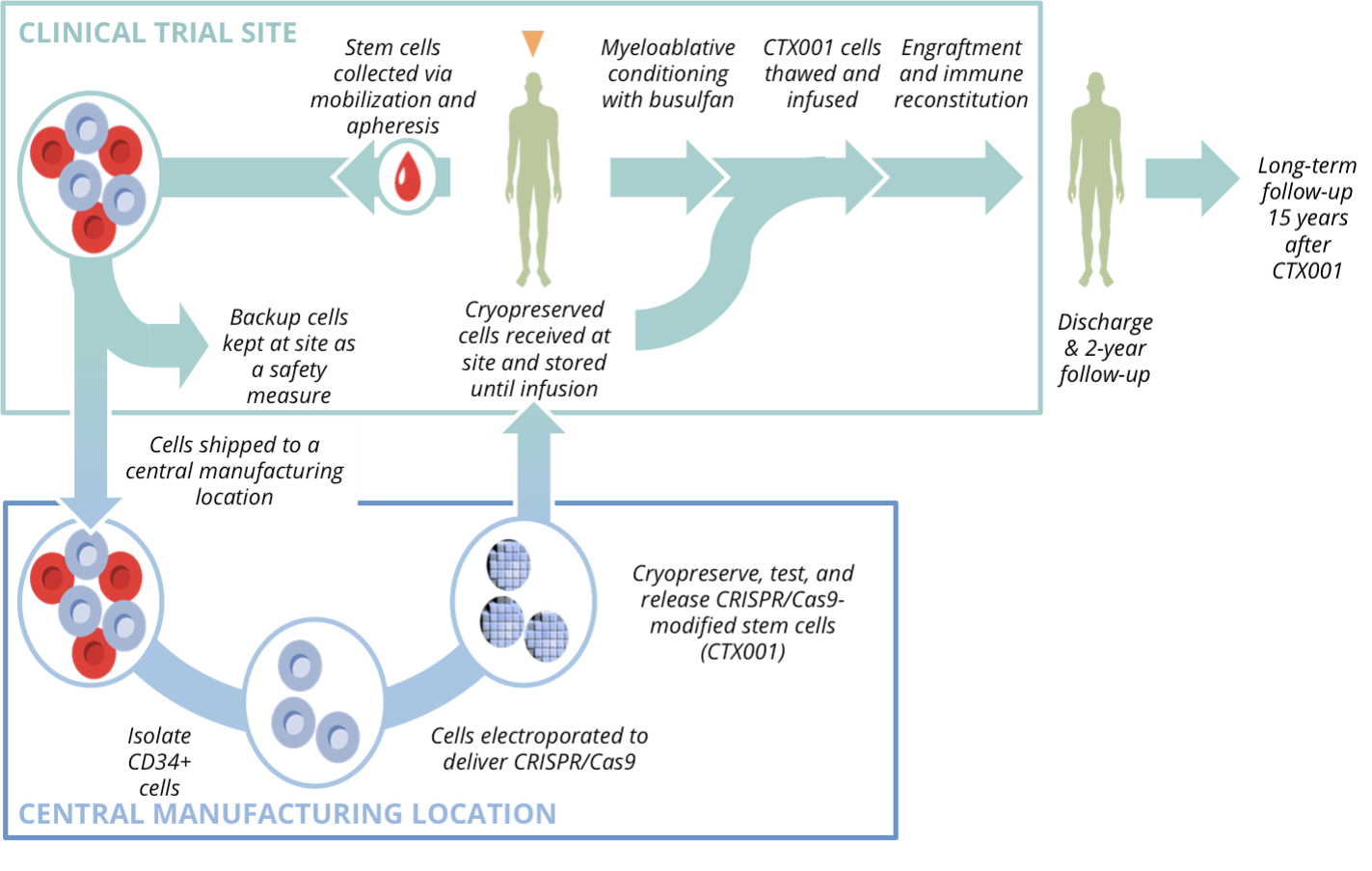
Schematic Representation of Manufacturing and Infusion Process
Evidence from the Trials
Clinical trials such as CLIMB-111 (for TDT) and CLIMB-121 (for SCD) have transformed theory into data:7,8
Sickle Cell Disease: About 90% of participants achieved complete freedom from vaso-occlusive crises for at least 12 months post-infusion.
β-Thalassemia: Approximately 89% attained transfusion independence for ≥12 months.
Sustained Outcomes: Most maintained total hemoglobin above 11 g/dL and HbF levels exceeding 20%.
These numbers are not just statistics — they represent lives untethered from blood transfusions and hospital corridors.
Safety and Unanswered Questions
Casgevy’s safety profile mirrors the intensity of its conditioning regimen. Myeloablation toxicity, infections, and transient cytopenias are the main risks. No CRISPR-related insertional mutagenesis or malignancy has been observed to date, reflecting the precision of non-integrating editing. Still, regulatory agencies mandate 15 years of follow-up to surveil for delayed adverse events, including clonal hematopoiesis or oncogenic transformation.9,10
As of 2025, the horizon remains reassuringly clear — but long-term genomic safety will define the legacy of this technology.
Who Can Receive Casgevy?
Casgevy is indicated for patients ≥12 years old who either:
Have sickle cell disease with recurrent vaso-occlusive crises, or
Have transfusion-dependent β-thalassemia without a suitable matched sibling donor.
The decision to proceed is multidisciplinary, involving hematologists, geneticists, and transplant specialists. Genetic counseling is essential, as the therapy’s benefits must be weighed against conditioning toxicity, infertility risk, and the need for extended hospitalization. Access remains limited to accredited gene-therapy centers.11,12
Ethics, Access, and the Future
Casgevy stands at the intersection of science and ethics. It exemplifies how genome editing can deliver functional cures, but also how innovation magnifies inequity. The therapy’s current cost — exceeding USD 2 million — underscores a broader challenge: how to democratize access to curative medicine. Efforts are underway in Australia, the UK, and the U.S. to create public–private frameworks for subsidized access through advanced therapeutics programs and health-technology assessment models.13,14
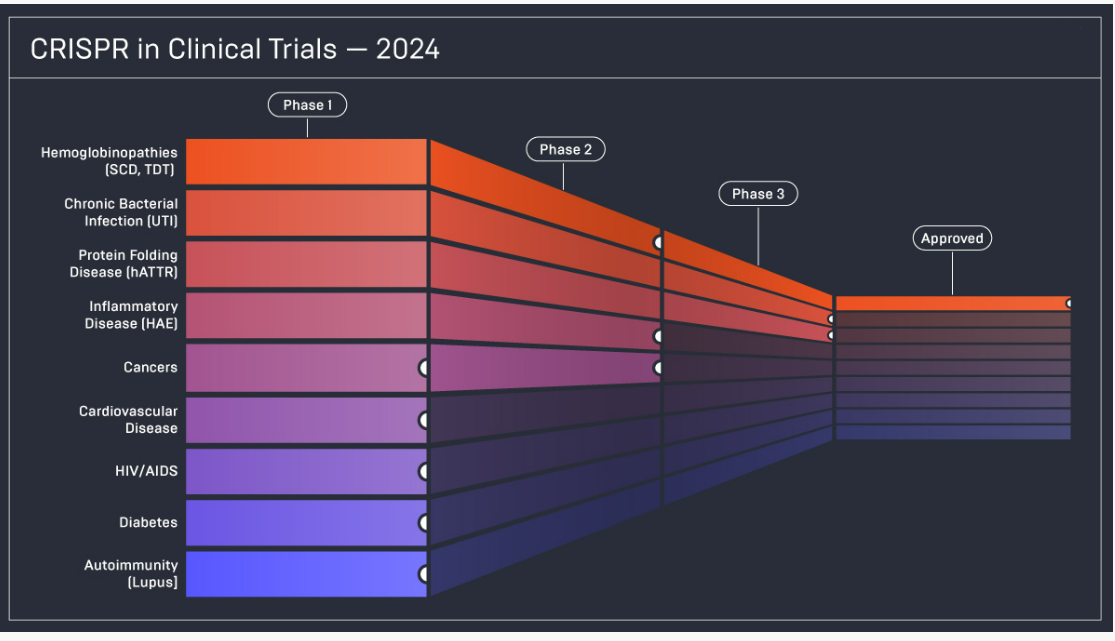
Beyond SCD and TDT, CRISPR-based editing is now being explored for hereditary angioedema, amyloidosis, and even HIV reservoir eradication — signaling a new genomic era in precision medicine.15,16
Conclusion
Casgevy represents far more than a therapeutic breakthrough; it marks a turning point in medicine itself. For the first time, clinicians are not merely countering the consequences of genetic disease but reshaping its underlying script. It stands as the first real-world demonstration that CRISPR-Cas9 can travel the full arc—from a bacterial defense mechanism, to a laboratory tool, to a treatment capable of transforming human lives.
What began as a curiosity of microbial immunity has matured into a precise instrument for rewriting dysfunctional biology. With Casgevy, genome editing steps out of theory and into clinical reality—ushering in an era where inherited disorders can be addressed at their genetic source, with the care, responsibility, and hope that such power demands.